Keywords
GBM; Hedgehog pathway; Wnt pathway; micro RNA
Introduction
Glioblastoma Multiforme (GBM) is the most aggressive primary glial neoplasm [1]. Because of its high potential for infiltration and invasion, local control of GBM is difficult with either surgery or radiation therapy. Surgical excisions as wide as hemispherectomy have failed to prevent tumor recurrence or improve survival [2,3]. Understanding and targeting of the invasion pathways has therefore, been focus of numerous experiments and pathological studies.
Invasive pathways involve complex interactions among cancer cells, extracellular matrix and white matter. These interactions are regulated by over 140 genes [4]. Multiple regulatory pathways including PI3K/Akt, Wnt, Sonic hedgehog and micro RNAs are implicated in the invasive properties of glioma cells [5]. There is significant genetic and phenotypic heterogeneity within the GBM [6-8]. Different subtypes of GBM such as pro neural (PN), neural, mesenchymal and classic also differ in their different genetic profile [9,10]. Understanding of invasive pathways and their heterogeneity is therefore important for targeting infiltrative potential of GBM. Our understanding of these pathways is, however, still incomplete.
The objective of this article was to review recent literature and provide an update on the basic mechanisms of local invasion of human GBM cells.
Literature Review
Scherer’s structures to the current understanding of invasion
Scherer, a German neuropathologist studied serial sections of the brain from 100 patients and concluded that glioma spreads along structures which he named “secondary structures” in 1938 [11]. These secondary structures, now called Scherer’s structures included brain parenchyma, preexisting blood vessels, white matter tracts and the subarachnoid space [11]. Another important observation that Scherer reported was that the invading cells do not have a single shape or form [11]. Rather, the shape of glioma cells varies according to the pathway they follow - this observation being consistent with the more recent hydrodynamic theory of invasion [12].
Migration of glioma cells mimics cellular migration during embryogenesis [12]. It starts with morphological polarization and development of membrane protrusions [5]. Glioma cells thus attach with the extracellular matrix (ECM) and pull themselves forward. The interaction of glioma cells with ECM is also chemical [5,13]. Several studies have shown tumor cells to influence stromal cells, altering the composition of the ECM [13]. Migratory cells undergo changes in volume and shape to allow movement through different spaces. The process is regulated by several growth factors, chemokines and cellextracellular matrix interactions. Degradation of the physical barrier of ECM by metalloproteinases (MMP) is essential for the migratory process. MMPs are shown to have a higher expression in glioma cells compared to their normal counterparts [5]. Several other enzymes including plasmin, cathepsin B, cathepsin D and heparanase help glioma cells evade ECM [14]. It has been shown that GBM cells demonstrate both mesenchymal migrations predominantly through Rac1 activation and amoeboid migration through Rho/Rhzo kinase (ROCK) activation [4,15,16].
GBM stem cells and invasive properties
GBM stem cells are a subpopulation of GBM cells that acquire the properties of a stem cell. These stem cell properties are important for their potential to infiltrate, recur and develop resistance to various therapeutic agents [17]. Stem cells in glioma are called glioma stem cells (GSCs) or glioma initiating cells (GICs). GSCs can be identified through their cell surface markers-CD 133 and Nestin [5]. Some GSCs can however, be CD133 negative [17,18]. GSCs capability of producing neurospheres in serum free culture conditions, selfrenewal and multipotency [19], contribute to their invasiveness of GBM and have attracted interest of scientists and pathologists for decades.
Glutamate promotes invasion
Glutamate acts in an autocrine and paracrine fashion to produces oscillatory Ca++ intracellular changes [12]. These changes are brought through Ca++-permeable AMPA receptors and promote invasion of glioma cells.
Wnt signaling pathway
Wingless/Int1 signaling pathways are involved in embryogenesis, tissue repair and cell self-renewal in adults [20]. Their mutations are implicated in several cancers including colon, breast, leukemias and GBM [20,21]. Wnt ligands Wnt1, Wnt 2, Wnt3a and Wnt5a are overexpressed in gliomas [22-24]. In mice, the knockdown of Wnt1 causes development of non-invasive small glial tumors while the knockdown of Wnt3 completely prevented the tumor formation [22]. And in vitro knockdown of Wnt5, suppresses MMP-2 and GBM invasion [23].
The three Wnt pathways are canonical Wnt/β-catenin dependent pathway, non-canonical dependent planar cell polarity pathway and the Wnt/Ca++ pathway. Of these three pathways, β-catenin dependent pathway is the most studied cascade [4,5,20].
β–catenin dependent pathway (Figure 1A) [25] starts when Wnt ligands bind with their seven-transmembrane cell surface receptor, called Frizzled (Fz). On binding Wnt factors, Fz interacts with its co-receptor low-density-lipoprotein-related protein 5/6 (LRP5/6), forming a complex that recruits the cytoplasmic scaffolding protein Dishevelled (Ds). The complex translocates to nucleus to interact with T-cell factor (TCF)/ lymphoid enhancer factor (LEF) transcription factors to regulate the expression of target genes. These target genes include c-Myc, cyclin D1, and MMPs [21,26]. C-Myc and cyclin D are involved in cellular proliferation while MMPs increase the invasive potential of glioma cells. Wnt factors that are known to activate this β-catenin dependent pathway are Wnt1, Wnt 3a and Wnt 7a [23]. β-catenin is overexpressed in glioma and its knockdown in vitro reduces invasiveness of GBM cells [24] (Figure 1).
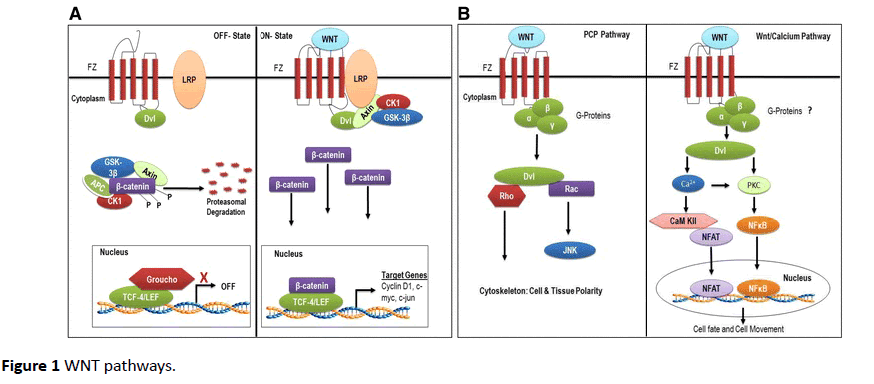
Figure 1 WNT pathways.
In non-canonical dependent (Figure 1B) [25] PCP pathway, Wnt signaling at Fz activates a mitogen activated protein kinase called Jun-N-terminal kinase. In Wnt/Ca++ pathway, various Wnt and Fz homologs activate calcium/calmodulindependent kinase II and protein kinase C [27]. These pathways have shown to be upregulated in GBM and are activated by Wnt2, Wnt4, Wnt5a, Wnt5b, Wnt6 and Wnt11 [23].
β-catenin degradation pathway
In the absence of Wnt signals, β –catenin binds with glycogen synthase kinase-3β (GSK-β) [21]. GSK-3β causes phosphorylation of β-catenin, which is then taken up by proteasome for degradation [16]. Several factors influence this pathway. EGFR trans-activates β-catenin by disrupting its association with α-catenin [28]. FRAT-1 prevents degradation of β-catenin by inhibiting GSK- β, thus upregulating GBM invasion and growth in vivo [29]. While, knockdown of Lef-1, a transcription factor inhibits GBM invasion in vitro [30].
PI3/Akt signaling pathway
PI3/Akt signaling pathway is implicated in up to 63% of grade III and IV tumors of the brain with an alteration in one of the four genes that form this pathway - epidermal growth factor receptor (EGFR), PTEN, PIK3R1 and PIK3CA [30]. The activation of PI3K pathway factors is associated with reduced survival [31], more aggressive behavior and treatment resistance [30]. PI3/Akt/mTOR pathway (Figure 2) [32] is activated by several growth factors like epithelial growth factor (EGF) and transforming growth factor-α (TGF-α). These factors upon binding receptors such as EGFR [32], gets homodimerized or heterdimerized, resulting in phosphorylation of its intracellular tyrosine kinase domain [33].
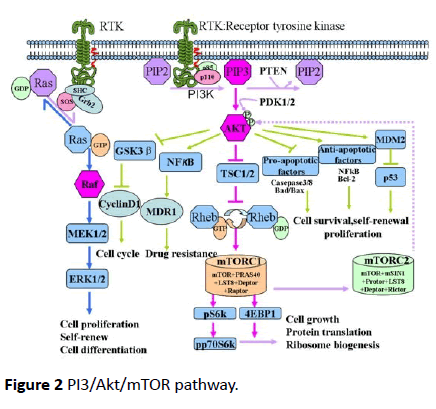
Figure 2 PI3/Akt/mTOR pathway.
Out of the three classes of PI3K, class I being the most vital to tumorigenesis, consists of a catalytic subunit p110 (α, β, γ) and a regulator subunit p85 [pI3oncotarget]. Upon activation phosphorylated tyrosine of EFGR binds to p85. The subsequent conformation change releases the catalytic subunit p110 [32]. The activated p110 phosphorylates the phosphatidylinositol- 3, 4-bisphosphate (PIP2) into the second messenger phosphatidylinositol-3, 4, 5-bisphosphate (PIP3) [15]. This reaction is reversed by the PI3K antagonist PTEN [34]. Subsequently, PIP3 will recruit the downstream Akt to inner membranes and phosphorylates Akt on its serine/threonine kinase sites (Thr308 and Ser473) [35,36]. Activated Akt phosphorylates several other proteins like GSK3β which stabilizes β-catenin [14] (Figure 2).
PI3/Akt increases MMP-2 and MMP-3 activity, especially in the peripheral cells enabling them to invade normal brain parenchyma [37]. PTEN on the other hand, suppresses MMP-2 and thus GBM invasion [34]. EGFR and PTEN mutation have been shown to cause continuous activation of PI3K/Akt/mTOR signaling pathway, thereby contributing to the tumorigenesis [32].
In PI3/Akt pathway, mTOR is an effector as well as upstream regulator [38,39]. mTOR resides in rapamycin-sensitive mTORcomplex (mTORC1) and a rapamycin-insensitive complex (mTORC2) [40,41]. mTORC2 phosphorylates Akt at Ser-473, taking part in metabolism, survival, proliferation and cytoskeletal reorganization of cells. Activated Akt inhibits tuberous sclerosis complex (TCS1/2), which in turn activates mTORC1. mTORC1 participates in protein translation, ribosomal biogenesis and cell growth [42,43].
Hedgehog-GLI1 pathway
Hedgehog pathways are critical to embryogenesis and tumorigenesis. Three types of ligands can activate hedgehog pathway i.e., sonic hedgehog (Shh), Indian hedgehog (ihh), and Desert hedgehog (Dhh) [44]. The receptor is a 12- transmembrane receptor Patched (PTCH). Binding ligand with the receptor abrogates another 7-transmembrane receptor protein called Smoothened (SMO) [44]. This pathway is shown in Figure 3 [45]. SMO prevents degradation of GLI1 which localizes to nucleus and upregulates several genes GLI1, PTCH1, cyclin D, Bcl-2 and VEGF [46,47]. GLI1 is also activated by other pathways like PI3/Akt and MEK. These pathways are activated by receptors tyrosine kinases like EGFR and PDGFR [48].
GLI family includes GLI1, GLI2, and GLI3 transcription factors. These factors act as effectors of Shh pathway. Another isoform of GLI1 called tGLI1 has been implicated in the invasive properties of GBM and breast cancers [49-52]. tGLI variant has not been detected in normal brain cells but is expressed in most GBM specimens [51,52]. tGLI has also been shown to upregulate the expression of VEGF-A and heparanase [52]. This leads to increased vascularity of GBM compared to the normal GLI variants [51]. Several studies have supported these observations [49-51]. Because of the lack of its expression in malignant cells, GLI can be an excellent target for therapeutic drugs (Figure 3).
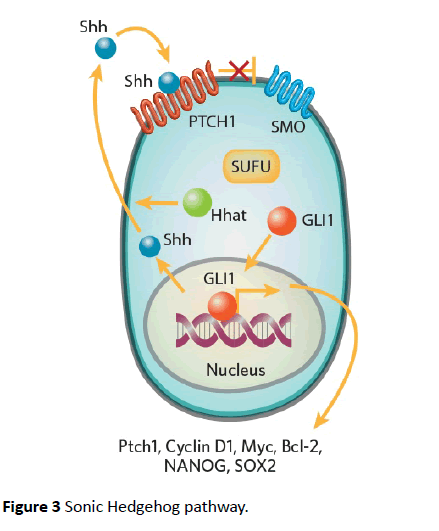
Figure 3 Sonic Hedgehog pathway.
The role of microRNAs
MicroRNAs (miRNA) are 20-25 nucleotide long, nonencoding RNA molecules that regulate the expression of more than 2/3rd of genes involved in stem cell related pathways [5]. They play an important role in the tumorigenesis and have been the focus of extensive research over the past decade. miRNAs bind to 3’untranslated regions (3’-UTRS), suppressing the translation of messenger RNA (mRNA) or inducing degradation of mRNA [38], affecting cell differentiation, proliferation and metabolism. Some of the miRNAs associated with invasion are discussed.
miR-218: miR-218 is significantly downregulated in GBM and correlates negatively with tumor grade [26,53,54]. It acts by downregulating Lef1, which is involved in the upregulation of β-catenin pathway. Lef1 downregulation cases reduction of MMP-2, MMP-7 and MMP-9, which inhibits cell invasion in vitro [26]. miR-218 also downregulates iKKβ mRNA and NF-kB [53]. NF-kB is involved in cellular invasion, increasing MMP-9 [5]. Other targets of miR-218 include GLI1, which is known to increase invasion of tumor cells [51,54,55].
miR-101: miR-101 is a tumor suppressor which is downregulated in GBM [56-58]. It acts on Kruppel-like factor 6 (KLF-6), reducing the expression of chitinase-3 like protein (CH13L1) and inactivating MEK1/2 and PI3 signaling [58]. KLF-6 is involved in cell invasion and migration, which gets inhibited as result of this downregulation.
miR-152: Another miRNA that inhbits invasion, apoptosis and migration of GBM stem cells (GMCs) and is downregulated in GBM [59]. Its acts by targeting Kruppel-like factor (KLF4) which downregulates galectin-3 (LGALS3) and inactivates MEK ½ an dPI3K signaling pathways [60].
miR-491: miR-491 is shown to inhibit the expression of protein kinase B (p-AKT1), proliferating cell nuclear antigen (PCNA), Cyclin D1 and MMP-2. This leads to arrest of GSCs in G0/G1 phase, reduced proliferative activity and invasive potential. This pathway is a target for new therapeutic agents [61].
miR-125b: miR-125 is an oncogene, commonly overexpressed in GSCs. Its suppression can decrease proliferative activity and invasive potential of GSCs [62]. It is also involved in the development of drug resistance to temozolamide (TMZ) [63]. An inhibition of the miR-125b on the other hand sensitizes GSCs to TMZ, downregulating MMP-2, MMP-9 through PIAS3 (which inhibits STAT signaling) in vivo and in vitro [64].
miR-181c: Overexpression of miR-181c inhibited GSC T98G migration and invasion [65,66]. The expression of E-cadherin was significantly upregulated, while N-cadherin and vimentin was significantly down-regulated by miR-181c [65]. miR-181c overexpression also inhibits TGF-β signaling by downregulating TGFBR1, TGFBR2 and TGFBRAP1 expression [65]. miR-181c thus plays a key role in GBM invasion, migration and mesenchymal transition [65,66].
Conclusion
GBM is an aggressive tumor with heterogeneous molecular features and complex host interactions involving the interplay of several pathways, many of which are amenable to therapeutic intervention.
23863
References
- John SY, Liu G, Ying H, Yong WH, Black KL, et al. (2004) Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Research 64: 4973-4979.
- Dandy WE (1928) Removal of right cerebral hemisphere for certain tumors with hemiplegia: Preliminary report. Journal of the American Medical Association 90: 823-825.
- Pang B, Wan W, Lee C, Khu KJ, Ng W (2007) The role of surgery in high-grade glioma-is surgical resection justified? A review of the current knowledge. Annals-Academy of Medicine Singapore 36: 358.
- Park JB, Agnihotri S, Golbourn B, Bertrand KC, Luck A, et al. Transcriptional profiling of GBM invasion genes identifies effective inhibitors of the LIM kinase-Cofilin pathway. Oncotarget 5: 9382.
- Paw I, Carpenter RC, Watabe K, Debinski W, Lo HW (2015) Mechanisms regulating glioma invasion. Cancer Letters 362: 1-7.
- Alves TR, Lima FRS, Kahn SA, Lobo D, Dubois LG, et al. (2011) Glioblastoma cells: A heterogeneous and fatal tumor interacting with the parenchyma. Life Sciences 89: 532-539.
- Rhee CH, Hess K, Jabbur J, Ruiz M, Yang Y, et al. (1999) cDNA expression array reveals heterogeneous gene expression profiles in three glioblastoma cell lines. Oncogene 18: 2711-2717.
- Inda MM, Bonavia R, Seoane J (2014) Glioblastoma multiforme: A look inside its heterogeneous nature. Cancers 6: 226-239.
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98-110.
- Network CGAR (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061.
- Scherer H (1938) Structural development in gliomas. The American Journal of Cancer 34: 333-351.
- Cuddapah VA, Robel S, Watkins S, Sontheimer H (2014) A neurocentric perspective on glioma invasion. Nature Reviews Neuroscience 15: 455.
- Bonnans C, Chou J, Werb Z (2014) Remodelling the extracellular matrix in development and disease. Nature Reviews Molecular Cell Biology 15: 786.
- Fonovi? M, Turk B (2014) Cysteine cathepsins and extracellular matrix degradation. Biochimicaet Biophysica Acta (BBA)-General Subjects 1840: 2560-2570.
- Frame MC, Brunton VG (2002) Advances in Rho-dependent actin regulation and oncogenic transformation. Current Opinion in Genetics & Development 12: 36-43.
- Weeks A, Okolowsky N, Golbourn B, Ivanchuk S, Smith C, et al. ECT2 and RASAL2 mediate mesenchymal-amoeboid transition in human astrocytoma cells. Am J Pathol 181: 662-674.
- Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, et al. (2007) CD133+ and CD133− glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Research 67: 4010-4015.
- Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, et al. (2010) A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 17: 362-375.
- Xie Q, Mittal S, Berens ME (2014) Targeting adaptive glioblastoma: An overview of proliferation and invasion. Neuro-oncology 16: 1575-1584.
- Clevers H (2006) Wnt/β-catenin signaling in development and disease. Cell 127: 469-480.
- Nager M, Bhardwaj D, Cantí C, Medina L, Nogués P, et al. (2012) β-Catenin signalling in glioblastoma multiforme and glioma-initiating cells. Chemotherapy Research and Practice.
- Kaur N, Chettiar S, Rathod S, Rath P, Muzumdar D, et al. (2013) Wnt3a mediated activation of Wnt/β-catenin signaling promotes tumor progression in glioblastoma. Molecular and Cellular Neuroscience 54: 44-57.
- Kamino M, Kishida M, Kibe T, Ikoma K, Iijima M, et al. (2011) Wnt?5a signaling is correlated with infiltrative activity in human glioma by inducing cellular migration and MMP?2. Cancer Science 102: 540-548.
- Pu P, Zhang Z, Kang C, Jiang R, Jia Z, et al. (2009) Downregulation of Wnt2 and [beta]-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Therapy 16: 351.
- Onyido EK, Sweeney E, Nateri AS (2016) Wnt-signalling pathways and microRNAs network in carcinogenesis: experimental and bioinformatics approaches. Molecular Cancer 15: 56.
- Liu Y, Yan W, Zhang W, Chen L, You G, et al. (2012) MiR-218 reverses high invasiveness of glioblastoma cells by targeting the oncogenic transcription factor LEF1. Oncology Reports 28: 1013-1021.
- Kohn AD, Moon RT (2005) Wnt and calcium signaling: β-catenin-independent pathways. Cell Calcium 38: 439-446.
- Gao X, Mi Y, Ma Y, Jin W (2014) LEF1 regulates glioblastoma cell proliferation, migration, invasion, and cancer stem-like cell self-renewal. Tumor Biology 35: 11505-11511.
- Guo G, Kuai D, Cai S, Xue N, Liu Y, et al. (2013) Knockdown of FRAT1 expression by RNA interference inhibits human glioblastoma cell growth, migration and invasion. PloS One 8: e61206.
- Kita D, Yonekawa Y, Weller M, Ohgaki H (2007) PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathologica 113: 295-302.
- Pelloski CE, Lin E, Zhang L, Yung WK, Colman H, et al. (2006) Prognostic associations of activated mitogen-activated protein kinase and Akt pathways in glioblastoma. Clinical Cancer Research 12: 3935-3941.
- Li X, Wu C, Chen N, Gu H, Yen A, et al. (2016) PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 7: 33440-33450.
- Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, et al. (2002) Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 110: 775-787.
- Koul D, Fu J, Shen R, LaFortune TA, Wang S, et al. (2012) Antitumor activity of NVP-BKM120—a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clinical Cancer Research 18: 184-195.
- Pitz MW, Eisenhauer EA, MacNeil MV, Thiessen B, Easaw JC, et al. (2015) Phase II study of PX-866 in recurrent glioblastoma. Neuro-Oncology 17: 1270-1274.
- Ma DJ, Galanis E, Anderson SK, Schiff D, Kaufmann TJ, et al. (2014) A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro-Oncology 17: 1261-1269.
- Kubiatowski T, Jang T, Lachyankar MB, Salmonsen R, Nabi RR, et al. (2001) Association of increased phosphatidylinositol 3-kinase signaling with increased invasiveness and gelatinase activity in malignant gliomas. J Neurosurg 95: 480-488.
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, et al. (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Research 66: 1500-1508.
- Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E (2014) PI3K/AKT signaling pathway and cancer: An updated review. Annals of Medicine 46: 372-383.
- Gulati N, Karsy M, Albert L, Murali R, Jhanwar-Uniyal M (2009) Involvement of mTORC1 and mTORC2 in regulation of glioblastoma multiforme growth and motility. Int J Oncol 35: 731-740.
- Wu SH, Bi JF, Cloughesy T, Cavenee WK, Mischel PS (2014) Emerging function of mTORC2 as a core regulator in glioblastoma: Metabolic reprogramming and drug resistance. Cancer Biology & Medicine 11: 255.
- Dowling RJ, Topisirovic I, Fonseca BD, Sonenberg N (2010) Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1804: 433-439.
- Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes & Development 18: 1926-1945.
- Clement V, Sanchez P, De Tribolet N, Radovanovic I, Altaba AR (2007) HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Current Biology 17: 165-172.
- Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW (2016) Targeting the Sonic Hedgehog signaling pathway: Review of Smoothened and GLI inhibitors. Cancers 8: 22.
- Bigelow RL, Chari NS, Undén AB, Spurgers KB, Lee S, et al. (2004) Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J Biol Chem 279: 1197-205.
- Zhu H, Lo HW (2010) The human glioma-associated oncogene homolog 1 (GLI1) family of transcription factors in gene regulation and diseases. Current Genomics 11: 238-245.
- Xie J, Aszterbaum M, Zhang X, Bonifas JM, Zachary C, et al. (2001) A role of PDGFRα in basal cell carcinoma proliferation. Proceedings of the National Academy of Sciences 98: 9255-9259.
- Cao X, Geradts J, Dewhirst M, Lo HW (2012) Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene 31: 104.
- Dikshit B, Irshad K, Madan E, Aggarwal N, Sarkar C, et al. (2013) FAT1 acts as an upstream regulator of oncogenic and inflammatory pathways, via PDCD4, in glioma cells. Oncogene 32: 3798.
- Lo HW, Zhu H, Cao X, Aldrich A, Ali-Osman F (2009) A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Research 69: 6790-6798.
- Zhu H, Carpenter RL, Han W, Lo HW (2014) The GLI1 splice variant TGLI1 promotes glioblastoma angiogenesis and growth. Cancer Letters 343: 51-61.
- Song L, Huang Q, Chen K, Liu L, Lin C, et al. (2010) miR-218 inhibits the invasive ability of glioma cells by direct downregulation of IKK-β. Biochemical and Biophysical Research Communications 402: 135-140.
- Peng B, Li D, Qin M, Luo D, Zhang X, et al. (2014) MicroRNA218 inhibits glioma migration and invasion via inhibiting glioma-associated oncogene homolog 1 expression at N terminus. Tumor Biology 35: 3831-3837.
- Tu Y, Gao X, Li G, Fu H, Cui D, et al. (2013) MicroRNA-218 inhibits glioma invasion, migration, proliferation, and cancer stem-like cell self-renewal by targeting the polycomb group gene Bmi1. Cancer Research 73: 6046-6055.
- Smits M, Nilsson J, Mir SE, Van Der Stoop PM, Hulleman E, et al. (2010) miR-101 is down-regulated in glioblastoma resulting in EZH2-induced proliferation, migration, and angiogenesis. Oncotarget 1: 710.
- Visani M, De Biase D, Marucci G, Cerasoli S, Nigrisoli E, et al. (2014) Expression of 19 microRNAs in glioblastoma and comparison with other brain neoplasia of grades I–III. Molecular Oncology 8: 417-430.
- Yao YL, Ma J, Wang P, Xue YX, Li Z, et al. (2015) miR?101 acts as a tumor suppressor by targeting Kruppel?like factor 6 in glioblastoma stem cells. CNS Neuroscience & Therapeutics 21: 40-51.
- Zheng X, Chopp M, Lu Y, Buller B, Jiang F (2013) MiR-15b and miR-152 reduce glioma cell invasion and angiogenesis via NRP-2 and MMP-3. Cancer Letters 329: 146-154.
- Mehran R, Nilsson M, Khajavi M, Du Z, Cascone T, et al. (2014) Tumor endothelial markers define novel subsets of cancer-specific circulating endothelial cells associated with antitumor efficacy. Cancer Research 74: 2731-2741.
- Pan S, Zhan S, Pei B, Sun Q, Bian L, et al. (2012) MicroRNA-149 inhibits proliferation and invasion of glioma cells via blockade of AKT1 signaling. Int J Immunopathol Pharmacol 25: 871-881.
- Shi L, Zhang J, Pan T, Zhou J, Gong W, et al. (2010) MiR-125b is critical for the suppression of human U251 glioma stem cell proliferation. Brain Research 1312: 120-126.
- Shi L, Zhang S, Feng K, Wu F, Wan Y, et al. (2012) MicroRNA-125b-2 confers human glioblastoma stem cells resistance to temozolomide through the mitochondrial pathway of apoptosis. International Journal of Oncology 40: 119-129.
- Shi L, Wan Y, Sun G, Zhang S, Wang Z, et al. (2014) miR-125b inhibitor may enhance the invasion-prevention activity of temozolomide in glioblastoma stem cells by targeting PIAS3. Bio Drugs 28: 41-54.
- He X, Liu Z, Peng Y, Yu C (2016) MicroRNA-181c inhibits glioblastoma cell invasion, migration and mesenchymal transition by targeting TGF-β pathway. Biochemical and Biophysical Research Communications 469: 1041-1048.
- Wang H, Tao T, Yan W, Feng Y, Wang Y, et al (2015) Upregulation of miR-181s reverses mesenchymal transition by targeting KPNA4 in glioblastoma. Scientific Reports p: 5.

